DNA分析揭示东亚松狮犬的起源


文 | 九屿
编辑 | 九屿
序言
动植物驯化是近代人类历史上最伟大的创新之一,是现代文明的根本基础;在所有适合驯化的大型哺乳动物中,只有少数物种成功驯化。
其中,家犬是唯一能够在人类创造的环境中茁壮成长的大型食肉动物,狗的驯化代表了人类组成的最迷人的进化过程之一。

尽管已经付出了广泛的努力来了解狗驯化的历史,但结论仍然存在很大争议;例如,mtDNA、Y染色体和全基因组测序指出东南亚是狗的起源地区。
使用SNP阵列对灰狼和家犬进行遗传比较表明,中东和中亚是狗驯化的重要来源;此外,一项古老的mtDNA工作也表明欧洲是狗驯化起源的另一个地点。
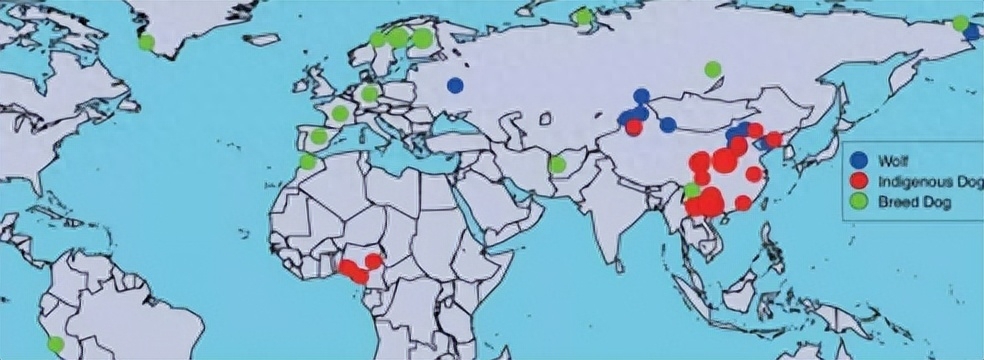

因此,欧亚大陆上的四个地理位置被认为是狗驯化的发源地,根据这些不同的研究,狗的起源似乎是一个很大的谜。
东亚有几种特有品种,包括工作犬、玩具犬以及气质和外表非常多样化的其他品种。
与过去几百年来通过强烈的人工选择的许多欧洲品种相比,东亚古代品种往往具有更多的遗传变异性,并且在形态上与欧洲品种一样独特。

东亚最有趣的古老品种之一是松狮犬,在中国历史上,松狮犬经常作为类似于佛教寺庙和宫殿前的传统石守护者的象征出现。它有一个结实的身材,外套非常致密,颈部特别厚。
此外,松狮犬还具有几个显着特征,包括额外的一对牙齿、不寻常的蓝黑色/紫色舌头和笔直的后腿,导致步态相当跷跷。

在这项研究中,我们对采样的松狮犬进行了限制性位点相关DNA测序。将这些序列与来自许多其他狗和亲戚的WGS数据相结合,我们根据东亚犬推断了松狮犬的起源,并确定了松狮犬谱系的适应性进化基因。
为了平衡测序成本和我们可以研究的个体数量,我们选择了RAD测序方法来调查松狮犬基因组。
在使用来自REBASE的所有II型限制性内切酶模拟狗参考基因组Canfam0.16中的切割位点后,我们选择了SpeI,它在狗参考基因组中有340,847个预测切割位点。
使用QIAamp DNA血液迷你试剂盒从QIAGEN中提取基因组DNA后,使用SpeI孵育基因组DNA16小时;将所得短片段连接到测序接头引物上;随后,对DNA片段进行超声处理以缩短片段。

在使用电泳进行尺寸选择后,我们使用来自Illumina的NEBNext Ultra DNA文库制备试剂盒来修复片段并将Y接头连接到超声处理的片段上。
配对的PCR引物与一个补充到P1衔接子,另一个包含条形码以及Y衔接子一个臂的互补序列,用于扩增靶基因组片段,其中一端具有P1衔接子,另一端具有Y衔接子。
我们还收集了19年发表的一只韩国珍岛犬的数据;我们总共收集了13种犬科动物的基因组序列,其中包括11只灰狼,1只中国本土犬,1只藏獒犬,48只松狮犬和其他犬种。
我们随机选择了14个E1区域和来自E11区域外的1个SNP来验证我们的SNP集,我们在八个松狮犬中对所有这些区域使用了Sanger测序。
为了计算SNP调用的假阳性和假阴性,我们首先确定每个Sanger读数的基因组区域;在桑格中找到但未在SNP集中发现的SNP被指定为假阴性;SNP 集专用但在 Sanger 测序中未找到的 SNP 被指定为假阳性。
为了比较不同狗和灰狼种群的连锁不平衡,我们将WGS数据与阵列数据相结合,提取了45,766个SNP,这些SNP在两个数据集中都进行了基因分型。

随后,我们选择了至少有九只个体的种群。
在从每个群体中随机选择500个个体后,使用PLINK计算彼此之间距离小于或等于29 kb的所有站点之间相关系数方面的连锁不平衡。

在处理每个总体后,我们将距离分箱为离散的500 kb窗口,并计算了所有不同大小窗口的平均r平方。为了测量连锁不平衡的总体水平,我们定义了一个H统计量,它是不同5 kb窗口的所有LD值的总和。
换句话说,H 统计量是kb 窗口内平均 LD 曲线下面积的模拟,并捕获每个总体中 LD 的总体水平。
首先,我们使用一系列过滤器来选择基因组中独立进化的中性位点;对于大多数SpeI切割地点,将对其相邻的上游和下游区域进行排序‘’我们选择了所有具有高覆盖率的SpeI切割位点。
切割位点两侧的200 bp序列连接在一起,随后从序列的两端修剪3 bp,在每个限制性内切酶切割位点产生0 bp位点。Canfm34.10中所有与CpG岛重叠的提取位点、重复区、间隙区也被移除。
为了将分析重点放在基因组的中性进化区域,我们保留了所有序列与外显子至少34 kb的距离,距离保守的非编码元件超过35 bp,并使用了来自UCSC和NCBI数据库的基因注释信息。
我们提取了狗基因组的CNE信息,类似于先前研究中描述的方法,使用位于UCSC的多物种比对的更新数据集。

由于G-PhoCS需要在整个基因组中独立进化的位点,因此我们每468 kb取一个位点。这些过滤器确定了200,可用于人口统计推断。
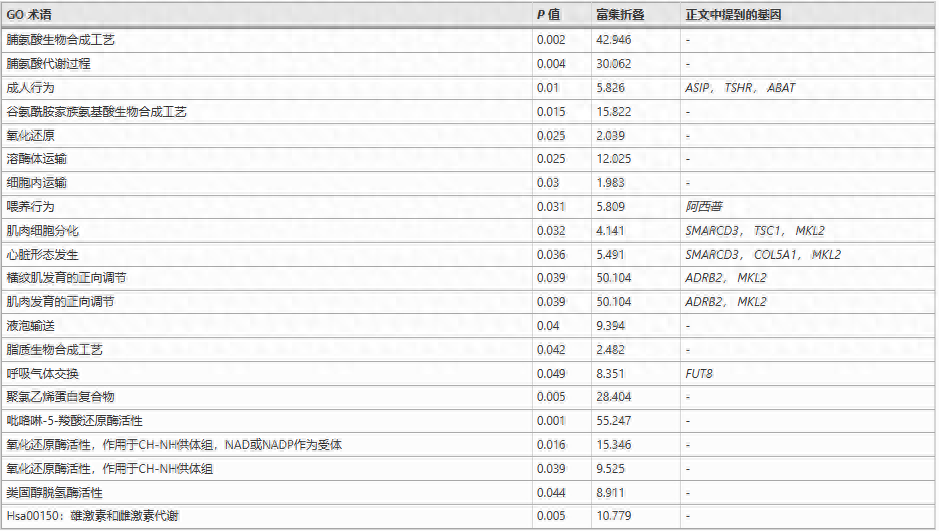
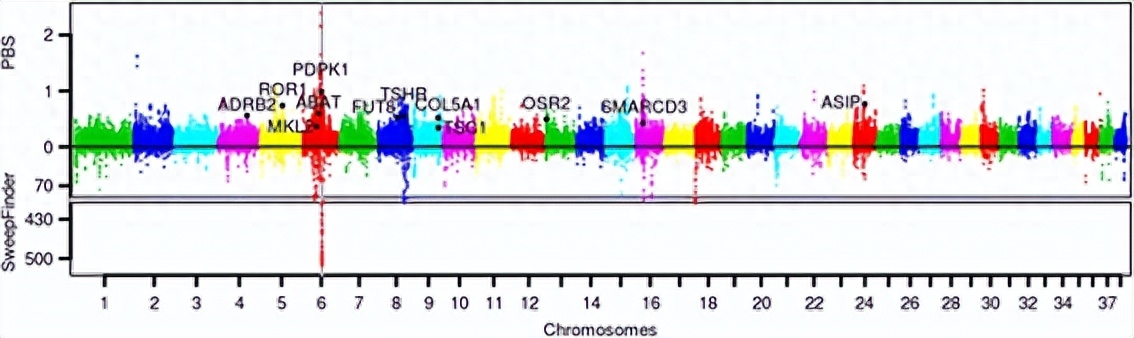
我们使用种群分支统计和SweepFinder,来识别松狮犬基因组中的选择性扫描区域;较高的PBS值代表沿松狮犬谱系的等位基因频率差异的长进化距离。

对于SweepFinder,我们使用dhole的基因组作为外群来识别所有SNP位置的祖先状态;然后,使用基因组背景位点频谱作为对照,使用SweepFinder作为识别选择性扫描痕迹的独立方法。
我们选择了PBS区域前3%和SweepFinder区域前3%的交集作为最终集合的候选区域,基因注释基于Ensembl注释。

之后,使用从Ensembl BioMart门户中提取的集合同源图谱,将这些狗基因ID转换为其相关的人类基因ID;使用DAVID进行基因本体分析。
为了探究松狮犬多余牙齿的遗传基础,我们进行了文献调查,发现四种重要途径参与牙齿发育。

随后,我们将所选基因列表与该基因列表重叠,寻找可能导致松狮犬牙齿数量不同的候选基因。对于松狮犬蓝舌的遗传基础,我们从颜色基因数据库中提取了所有色素沉着基因。
使用改进的RAD构建方案,使用Illumina平台汇集和测序来自1个个体的测序文库。在RAD测序中,配对末端测序的一端严格定位在同一限制性切割部位并具有均匀的覆盖范围,而另一端的位置根据插入片段尺寸而变化。

我们的RAD测序生成的序列数据,与由来自四项已发表研究的37种犬科动物的全基因组序列组成的数据集相结合;我们总共在整个基因组中调用了16,716,649个SNP。
该集合的转换/转换比为2.186,表明SAMtools中实现的变体调用过程的结果质量良好;我们将这个SNP集表示为全基因组SNP集。

由于RAD测序的序列覆盖率将仅限于某些基因组区域,我们通过靶向RAD个体具有良好覆盖率的基因组区域来进一步过滤SNP集并提取了910,5,2个高质量SNP。
我们发现,RAD数据中变体调用中的假阳性和假阴性分别为6.4%和1.2%。随后的群体遗传分析是使用这两个SNP集的不同组合进行的。

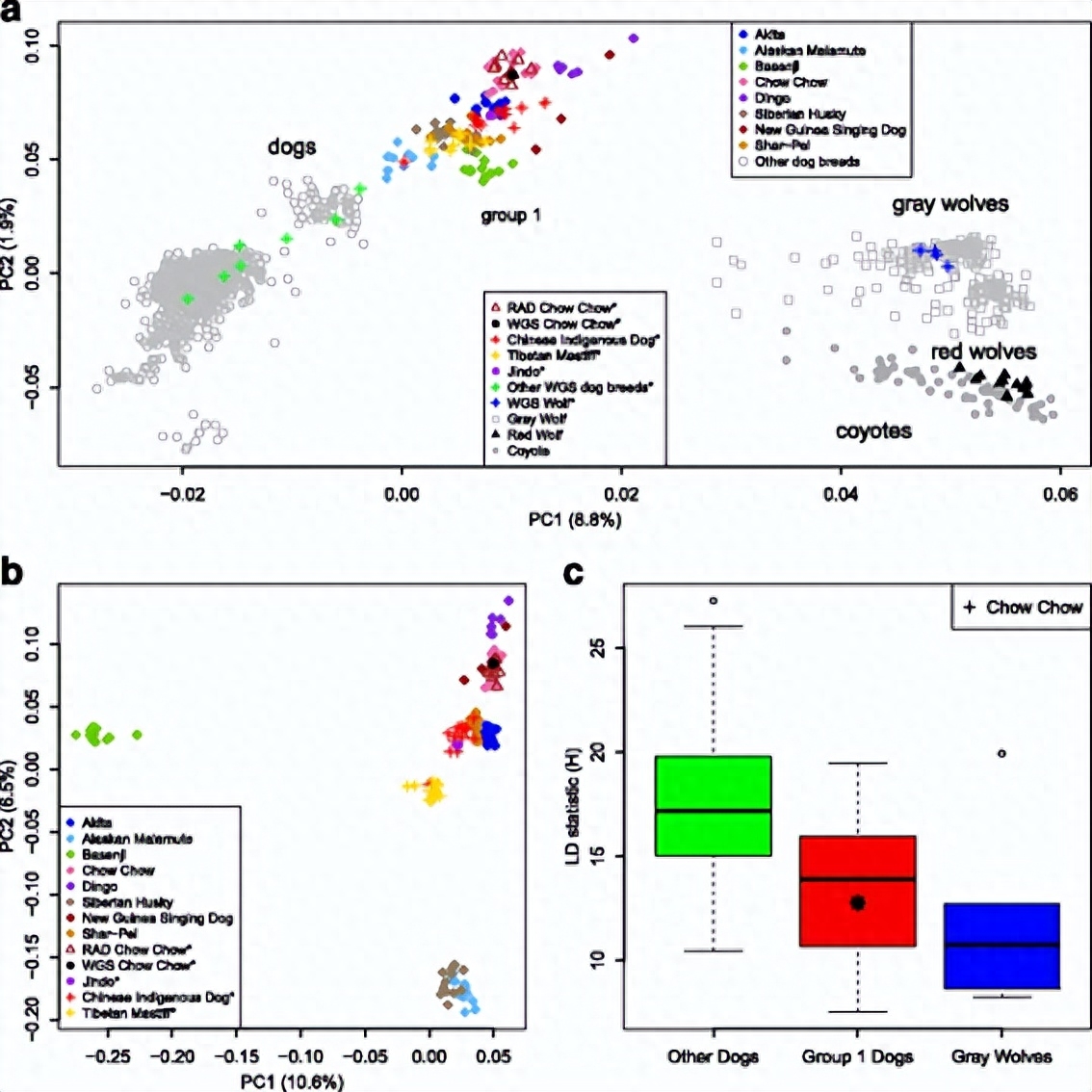
使用亲属关系估计程序,我们发现我们收集的松狮犬并不密切相关,并且与SNP阵列中的个体显示出相似的亲属价值。
相当令人欣慰的是,松狮犬的多个来源在PCA图中彼此一致,这表明为松狮犬收集的不同数据集中的样本质量非常一致。

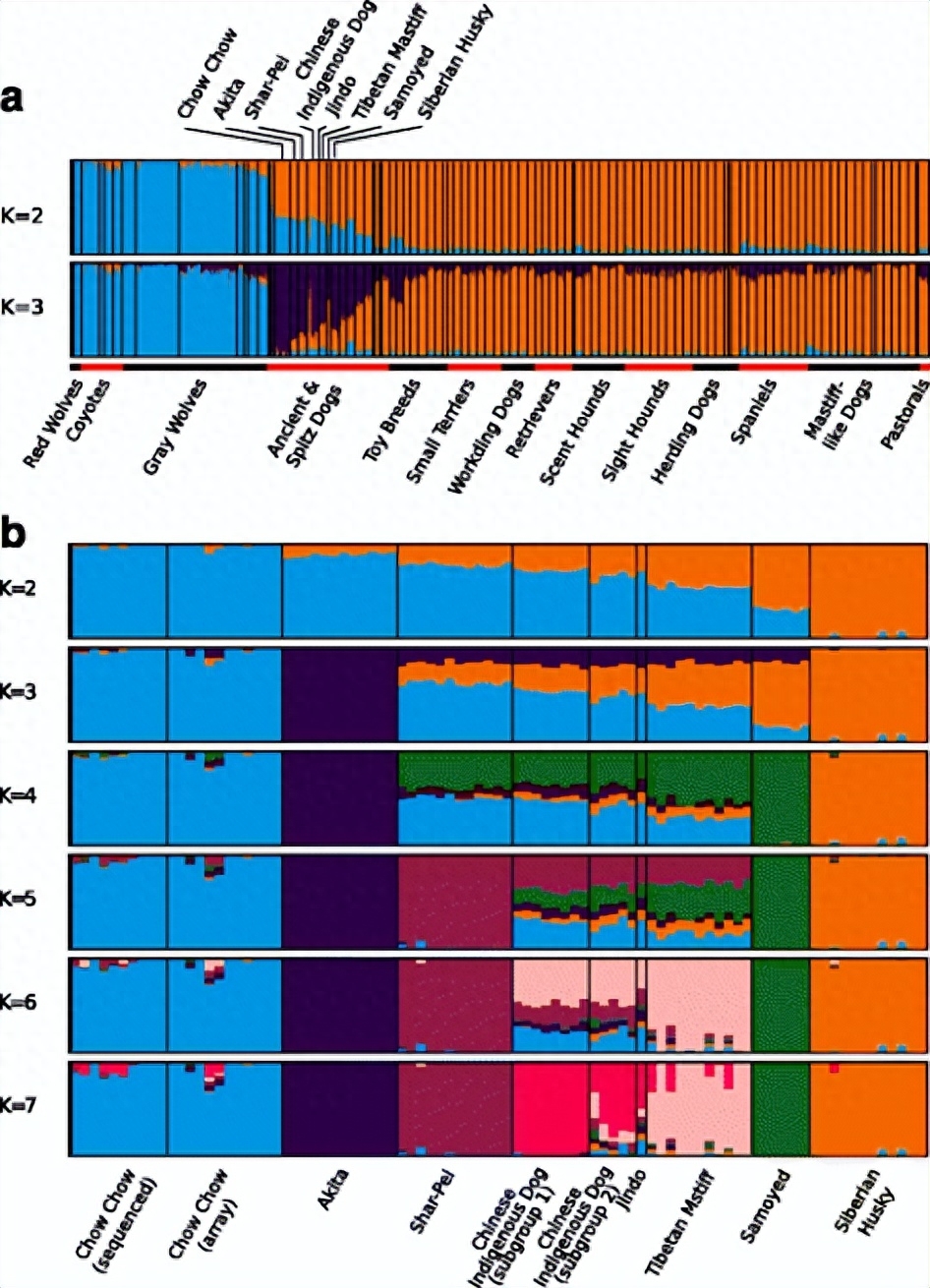
为了进一步剖析第1组个体之间的关系,对这些个体进行了PCA分析我们发现聚类模式与这些个体的地理起源密切相关。
例如,巴辛吉与沿第一轴的其余个体保持不同。随后,来自北极地区的品种沿 PC1 轴与其他品种分开,其余的东亚品种彼此保持接近。

由于一个起源于中国的古老品种,产生松狮犬的时间和过程应该有助于我们了解东亚的品种形成。使用基于多个序列之间的差异和良好校准的突变率的马尔可夫链蒙特卡罗方法,我们确定了松狮犬起源于中国本土犬的年代。
我们发现狗在大约31,700年前与东亚的灰狼分离,与早期的几个发现相匹配;这两个种群分离后,祖先中国本土犬保持了相对较少的种群。

松狮犬起源于中国本土犬的时间估计为8300年前;有趣的是,从松狮犬分离出来后,中国本土犬的数量规模迅速增加,而松狮犬的整体种群规模与祖先中国本土犬的种群规模相比略有下降。
Ror1和Ror2基因均在小鼠磨牙原基中表达,Ror2小鼠的牙齿分化缺陷,缺乏Osr2基因的小鼠在臼齿舌侧发育出多余的牙齿。
以松狮犬为示例品种,我们对一种古老的东亚犬种进行了系统的研究;有几个有趣的观察结果值得在这里讨论。
首先,东亚品种的起源时间可能相当古老,松狮犬是最接近中国本土犬的品种;如果所有其他犬种也都起源于中国本土犬,那么它们一定都起源于8300年前。

导致松狮犬的温和种群瓶颈表明,这是一个逐渐的过程,导致该品种的形成;从历史上看,根据密集外套的形态特征,松狮犬经常被认为是高纬度起源的品种。
然而,这里的分析表明,松狮犬是从中国本土犬中挑选出来的,这些犬是南方血统的。
鉴于农业始于大约11,000至9000年前在长江附近的东亚,人类久坐的环境可能促进了从中国本土狗中选择松狮犬。
其次,尽管松狮犬的出现被假设是渐进的,但中国本土犬和松狮犬之间的整体基因流动量却低得惊人。当一个早期品种正在发育时,源种群和感兴趣的种群之间的遗传交换量可能相当高,因为分化量仍然很低。

第三,我们使用基因本体的基于基因的分析仅符合松狮犬特有的表型期望的子集;例如,GO分析结果中没有强烈指出蓝色舌头和厚外套。
对此的一个可能的解释是,负责这些表型的基因可能非常简单,并且不容易被基于GO的分析所拾取。

人口学推断发现,松狮犬起源于8300年前的中国本土犬,导致松狮犬的进化过程伴随着低水平的基因流动和轻微的种群瓶颈。
两类基因显示出沿着松狮犬谱系的正向选择的有力证据,即与代谢和消化相关的基因以及与肌肉/心脏发育和分化相关的基因,对松狮犬的研究为东亚品种产生的历史和过程提供了重要的见解。


















 鲁公网安备37020202001850号
鲁公网安备37020202001850号